Publications
Teratology Primer, 3rd Edition
What Are the Ethical and Scientific Considerations for the Inclusion of Pregnant and Breastfeeding Women in Clinical Trials?
Leyla Sahin, MD, FACOG FDA Center for Drug Evaluation and Research, Office of New Drugs Division of Pediatric and Maternal Health
This chapter represents the opinions of the author, and does not necessarily represent the official position of the FDA.
Pregnant and breastfeeding women can become ill or have chronic medical conditions that require treatment. Therefore, information on drug safety, effectiveness and dosing in pregnancy, and safety information for infants who may get exposed through lactation is important. The majority of pre-approval clinical trials performed to evaluate the dosing, safety, and efficacy of pharmaceuticals are conducted in adult, nonpregnant subjects. Pregnant and breastfeeding women are often excluded from these trials to protect the fetus or infant from risks associated with exposure to an investigational (test) product. The scientific and ethical complexities of enrolling pregnant women in research are challenging. While these challenges exist, when it is clear that pregnant and breastfeeding women will use a drug after it is approved, there is a clear public health need to understand the safety and efficacy of medications used during pregnancy and lactation. To address this need for better information, federal agencies, academia, and other stakeholders and advocates have been working to increase the inclusion of pregnant and breastfeeding women in clinical trials that are conducted according to scientific and ethical guidelines.
The need for data to inform prescribing in these populations raises the question regarding which type of study is the most appropriate. In considering research in pregnant and lactating women, it is helpful to distinguish clinical studies from observational studies. Furthermore, in considering clinical studies, it is helpful to distinguish interventional studies from opportunistic studies (see Table 1).
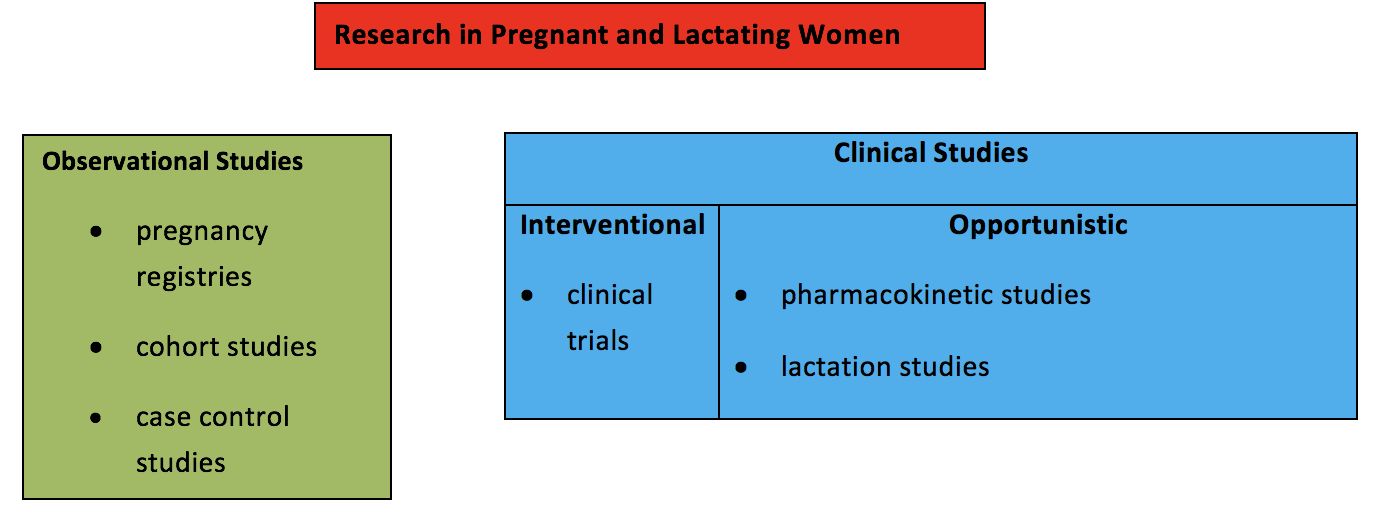
Observational studies assess the safety of medications in pregnancy and are conducted after a drug has been marketed. Observational studies involve evaluation of information already accruing in the post-marketing environment as a result of clinical practice decisions. Clinical studies, such as clinical trials where a study drug is administered, involve potential risk to the pregnant woman and the fetus. Opportunistic clinical studies enroll pregnant or breastfeeding women who are already taking a medication that was prescribed by their health care providers. A medication is not given to the pregnant woman as part of the study, and there is no comparison to a placebo or treatment arm. Studies that enroll women who are already receiving a medication that was prescribed by their own health care providers for treatment of their medical condition are generally considered minimal risk. The only study-related risks include blood draws and expression of milk using a breast pump, which are considered minimal risk. Most lactation studies that evaluate how much medication passes into breast milk fall into this category. Studies that report pharmacokinetic data collected during pregnancy to determine whether dose adjustments are need in pregnant women who are receiving a medication prescribed by their health care provider also fall into this category.
All human research is governed by current federal regulations, referred to as the “Common Rule” which provide for human subject protection to ensure that research participants are not exploited, are respected for their ability to make informed choices, and are provided accurate information that reflects the potential risks and benefits of the study. The general ethical framework ensures that the research has scientific value by addressing an unmet need and validity by being scientifically rigorous and interpretable. The regulations require that an Institutional Review Board (IRB) provide independent ethical and scientific oversight to a clinical study, serving as the gate-keeper in protecting the rights and welfare of study participants. Additionally, there are other regulations that specifically address pregnant women, referred to as “Subpart B”. These regulations have the following 10 requirements that need to be met in order to include pregnant women in clinical trials supported or conducted by the Department of Health and Human Services:
- Where scientifically appropriate, preclinical studies, including studies on pregnant animals, and clinical studies, including studies on nonpregnant women, have been conducted and provide data for assessing potential risks to pregnant women and fetuses.
- The study must provide potential benefit for the woman or the fetus. If there is no such prospect of benefit, there must be minimal risk to the fetus, and the purpose of the research is the development of important biomedical knowledge which cannot be obtained by any other means.
- Any risk is the least possible for achieving the objectives of the research.
- The pregnant woman's consent is obtained.
- If the research is only for the purpose of benefiting the fetus, the consent of the father is required as well, with the following exceptions: if he is unable to consent because of unavailability, incompetence, or temporary incapacity or the pregnancy resulted from rape or incest.
- Each individual providing consent is fully informed regarding the reasonably foreseeable impact of the research on the fetus or neonate.
- For minors who are pregnant, consent is obtained in accordance with local state laws.
- No inducements, monetary or otherwise, will be offered to terminate a pregnancy.
- Individuals engaged in the research will have no part in any decisions as to the timing, method, or procedures used to terminate a pregnancy.
- Individuals engaged in the research will have no part in determining the viability of a neonate.
FDA recommends that the 10 requirements above be satisfied for FDA-regulated clinical research.
In considering when it would be ethically justifiable to include pregnant women in clinical trials, it is helpful to consider the postmarketing setting (i.e., FDA approved drugs) separately from the premarketing setting (i.e. an investigational drug that is not FDA approved). For approved drugs, there may be a need to determine efficacy in pregnancy due to concerns regarding physiologic or metabolic changes that occur as the pregnancy progresses. For example, randomized controlled trials (RCTs) have been conducted to assess the effectiveness and safety of oral hypoglycemic agents in the management of gestational diabetes. There may also be a need to assess a safety outcome that is specific to pregnancy. For example, RCTs have been conducted to assess the incidence and severity of neonatal withdrawal syndrome in opioid-dependent pregnant women on methadone compared with buprenorphine. As illustrated by these examples, it would be ethically and scientifically acceptable to conduct RCTs in pregnant women for approved drugs if the requirements, as outlined in Subpart B (above) are met: efficacy and/or safety cannot be assessed by other less risky study methods, adequate preclinical studies (including studies on pregnant animals) have been completed, and there is an established safety database in nonpregnant women from clinical trials or preliminary safety data from the medical literature and/or other sources regarding use in pregnant women.
Inclusion of pregnant women in clinical trials of premarket investigational products involves greater potential risk, because available safety and efficacy data are limited. Therefore there are additional risk-benefit and ethical considerations and safeguards for research participants. Questions that need to be addressed include whether the potential benefit outweighs the potential risk? Is it a life-threatening condition for which there is no other treatment? Is there no other way to obtain the data? Considerations that may make it ethically justifiable to include pregnant women in these types of clinical trials include the study of treatments for a serious or life-threatening condition for which there are no other available effective therapies (e.g., endemic infections), and the development of drugs to treat pregnancy-related conditions. Pregnant and breastfeeding women are populations that have not been well studied historically. However, these populations deserve access to medications that have been adequately evaluated, and can be studied in an ethically and scientifically sound manner.
Suggested Reading
The Common Rule 45 CFR 46 Part A
https://www.hhs.gov/ohrp/regulations-and-policy/regulations/45-cfr-46/index.html
Additional Protections for Pregnant Women 45 CFR 46, Part B
https://www.hhs.gov/ohrp/regulations-and-policy/regulations/45-cfr-46/index.html#subpartb
Balsells M, García-Patterson A, Solà I, et al. Glibenclamide, metformin, and insulin for the treatment of gestational diabetes: a systematic review and meta-analysis. BMJ 2015 Jan 21.
Jones HE, Kaltenbach K, Heil SH, et al. Neonatal abstinence syndrome after methadone or buprenorphine exposure. N Engl J Med. 2010 Dec 9; 363(24):2320-31.