Publications
Teratology Primer, 3rd Edition
How are Prescription Medications Labeled for Pregnancy and Lactation?
Leyla Sahin, MD, FACOG FDA, Center for Drug Evaluation and Research, Office of New Drugs, Division of Pediatric and Maternal Health
Marie Teil, MD UCB Pharma, Immunology Patient Value Unit, Women of Childbearing Age and Pediatrics Program
This chapter represents the opinions of the author, and does not necessarily represent the official position of the FDA or UCB.
Prescription medication labeling (also known as prescribing information and package insert) in the US, or Summary of Product Characteristics in the EU, is a compilation of information about a product that is written by the manufacturer and approved by the Food and Drug Administration (FDA), or European Medicines Agency (EMA). Labeling is based on the regulatory authority's review of the new drug application (NDA) or biological license application (BLA), and includes information necessary for safe and effective use. Updates are made as new information becomes available. Labeling is written primarily for the health care provider.
Women of childbearing potential affected with chronic diseases may require continuation of prescription medicines before, during and after pregnancy to ensure the health of the mother and the baby. Women may also develop acute conditions during pregnancy that need treatment. Healthcare professionals may face challenging situations such as prescription of medicinal products to women of child-bearing potential, pregnant or breast-feeding women. The product information on pregnancy and lactation is important to provide relevant information on the use of medicinal products in human reproduction, pregnancy and breastfeeding.
The information included in the pregnancy and lactation sections of prescription product labeling is based on a comprehensive review of all available data, including nonclinical (animal) reproductive and developmental toxicity data, and human data. During drug development and the early post marketing period non-clinical data are of great importance since clinical experience is still lacking or limited, resulting in little if any information on the use of the drug in pregnant or lactating women.
Clinical data becomes more important as experience grows during the life cycle of the product, and companies are required to incorporate clinically relevant data into existing labeling. Such data may be derived from post-marketing surveillance (either voluntary or required), observational studies, published literature, clinical trials, and pharmacokinetic or lactation studies.
FDA and EMA perform a multi-disciplinary assessment of all available data to assess the quality of evidence. Data are added to labeling if they are determined to be meaningful or clinically relevant for the healthcare provider. Differences between FDA and EMA approaches in label outcomes are described below.
US Labeling
Before a new prescription medication is approved by the FDA, product labeling must be developed. Prescription product labeling is a detailed document that must include information about the approved use(s) of the drug.
FDA-approved product labeling is accessible electronically and is an important tool for prescribers in communicating risks, benefits, and uncertainties to their patients. In the United States, prescription drugs and biological products must include information on the safety of the drug when used during pregnancy or lactation. On June 30, 2015, FDA began implementing its new pregnancy and lactation labeling system, which eliminates the pregnancy letter categories (A, B, C, D, and X), and replaces this information with narrative summaries of available data. These changes are occurring through the year 2020, and labeling will be further updated as new information becomes available. The pregnancy letter categories are being eliminated because they were overly simplistic and did not capture the complexities and nuances of available risk information and benefit-risk considerations. The pregnancy letter categories were misinterpreted as a grading system, which sometimes resulted in misguided prescribing decisions. The new labeling system provides clinically relevant information that presents the background risk of birth defects and miscarriage for context, and, when applicable, the adverse pregnancy outcomes associated with the underlying medical condition.
Under the new labeling system, Section 8 – USE IN SPECIFIC POPULATIONS has subsections for pregnancy, lactation and reproductive potential (Figure 1). The Pregnancy (8.1) and Lactation (8.2) subsections include three headings: “Risk summary,” “Clinical Considerations” and “Data.” These headings provide information regarding human, animal, and pharmacologic data on the use of the medication, and adverse reactions of concern for pregnant or breastfeeding women. If applicable, the Pregnancy subsection also includes the contact information for a pregnancy exposure registry that is enrolling pregnant women exposed to the medication.
Figure 1. Overview of US Pregnancy and Lactation Labeling.
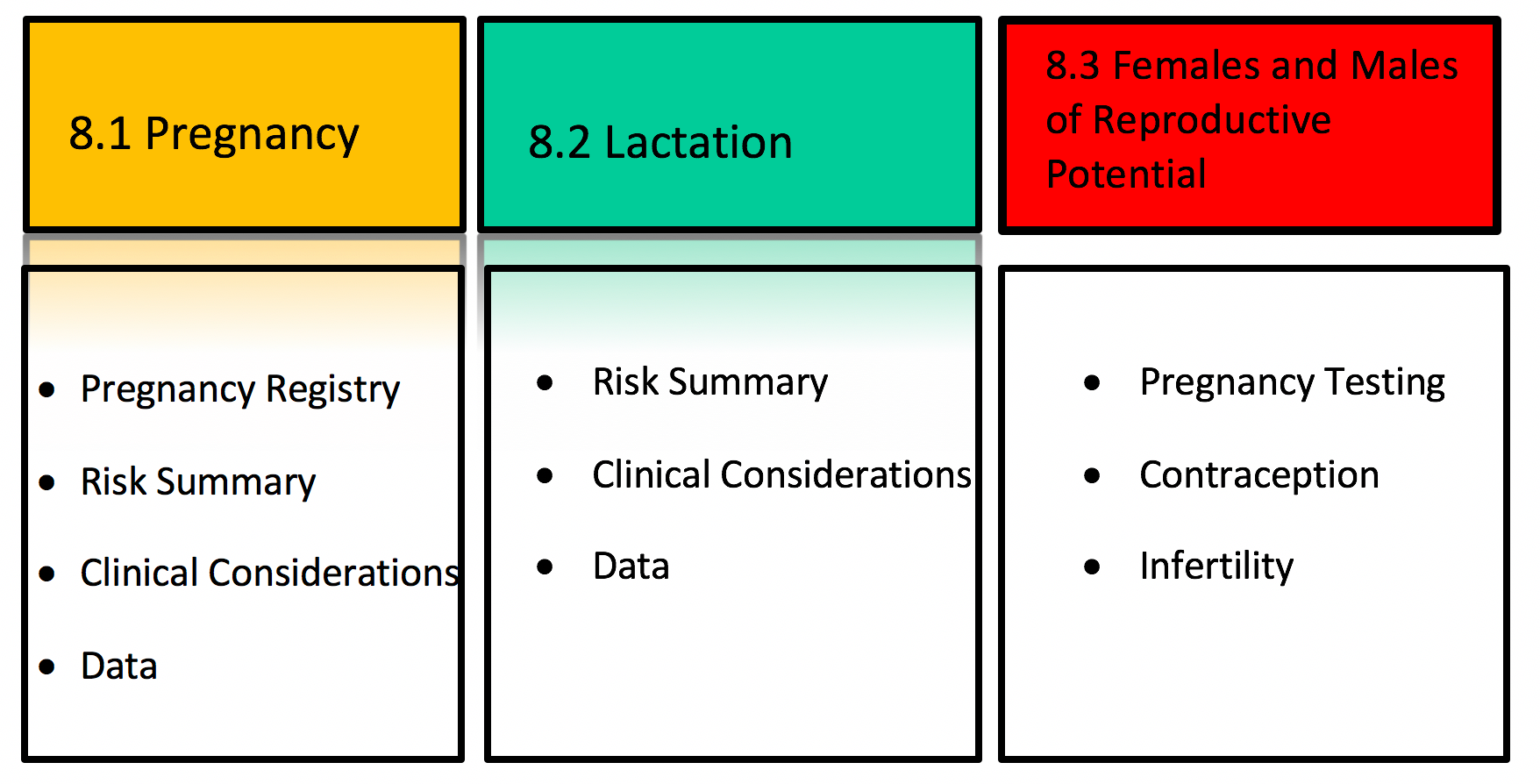
The Risk Summary is a narrative summary of the data, and conveys what is known about the potential risk of exposure for the medication during pregnancy. Additionally, the Risk Summary includes the background rate for major birth defects and miscarriage in the general population, regardless of medication exposure. The absence of data to inform risk is conveyed if appropriate.
The Clinical Considerations heading under Pregnancy includes additional information relevant to the use of a medication in pregnancy. Information may include possible impacts of untreated disease in order to provide balanced risk-benefit information. For example, poorly controlled Crohn’s disease in pregnancy is associated with adverse pregnancy outcomes, including fetal loss, preterm delivery, and small for gestational age. The Clinical Considerations section also includes the subheading, “Dose adjustments during pregnancy and the postpartum period,” which presents clinical pharmacology information that inform dosing during pregnancy and the postpartum period.
The Data section includes a concise description of the studies, both human and animal, that support the statements in the Risk Summary.
The Lactation subsection (8.2) provides information about using the medication while breastfeeding, such as the amount of medication in breast milk and potential effects on the breastfed infant. The Clinical Considerations heading under Lactation includes information on minimizing exposure to the breastfed infant, if applicable.
The Females and Males of Reproductive Potential subsection (8.3) presents information about pregnancy testing and contraception recommendations for medications with suspected or known adverse pregnancy effects and information about infertility effects.
The new labeling system for medications in pregnancy and lactation is an important step in increasing the availability of clear and current information for prescribers, pregnant women, and breastfeeding women.
EMA Labeling
In the EU, the Summary of Product Characteristic (SmPC) is the key reference document on a medicinal product and the basis for information on how to use this medicinal product safely and effectively. The SmPC is the result of the agreed position on the medicinal product, as defined during the assessment process, before and after marketing authorization. It contributes to informing on factors that may influence the benefits and the risks. The SmPC is updated throughout the life of the product as new data are collected and is available to healthcare professionals, patients, and consumers (organizations). The content and the order of the SmPC are defined by Article 11 of Directive 2001/83/EC. Section 4.6 entitled Fertility, Pregnancy and Lactation provides information on the use of a medicine product in relation to reproduction. More specifically, this section refers to contraception, pregnancy, breastfeeding, and fertility.
The Committee for Medicinal Products for Human Use (CHMP) guideline on risk assessment of medicinal products on human reproduction and lactation from data to labeling (EMEA/CHMP/203927/2005) describes the process of assessment of available data and the integration of animal and human data to evaluate the risks of the use of medicinal products during pregnancy. The assessment aims at avoiding an increased risk of birth defects compared to the untreated population.
The Non-Clinical Assessment is based on non-clinical pharmacological and pharmacokinetic properties of the medicinal product, results from non-clinical toxicity studies, reproductive toxicity studies, and all available pharmacological and toxicological data. Toxicity studies can provide important information regarding potential effects on reproduction, including male fertility. Conclusions of the non-clinical assessment are based on the detection/lack of detection of a suggested reproductive toxicity effect.
The Clinical Assessment is based on comprehensive information on human data such as the extent of the clinical post marketing experience and any additional results from clinical studies if available.
- The Women of Childbearing Potential sub-section provide information on the use of contraceptive measures (in males and females): The assessment is based on product molecular structure, mode of action, and pre-clinical and clinical study results including post marketing experience. Recommendations on the use of the medicinal product in women of childbearing potential may include pregnancy testing and contraception language, including defined period before starting and/or after ending treatment (wash-out period).
- The Pregnancy sub-section includes conclusions from animal studies and human data if available and an overall recommendation on the use of the medicinal product during pregnancy. The recommendation is based on the integration of the pre-clinical assessment conclusions (i.e. effect detected vs. effect not detected) together with the conclusion of the clinical assessment (i.e. demonstrated or suspected human teratogenicity or fetotoxicity; no increased incidence of malformations observed within a moderate amount (at least 300) of 1st trimester prospectively exposed pregnancies with known outcome; no increased incidence of malformations observed within a large amount (at least 1000) of 1st trimester prospectively exposed pregnancies with known outcome). The outcome is a recommendation on the use of the medicine during different periods of gestation with or without recommendations on the management of pregnancy based upon timing of exposure.
- The Lactation sub-section provides available data if any and a recommendation based on all available data including pre-clinical results and potential transfer into breast milk.
- The Fertility section includes all known data if available from both male and female fertility studies, based on the characteristics of the medicinal product, as identified in the non-clinical studies and/or due to the pharmacological profile
Additional collection and analysis of human data during the lifecycle of the medicinal product, and their inclusion in section 4.6, are essential to continual provision of the most current information for healthcare providers, pregnant, and breastfeeding women.
Suggested Reading
Labeling Information for Drug Products Webpage https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/default.htm
Pregnancy and Lactation Labeling Rule Webpage https://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/labeling/ucm093307.htm
Summary of Product Characteristic https://ec.europa.eu/health/sites/health/files/files/eudralex/vol1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
The Committee for Medicinal Products for Human Use guideline on risk assessment of medicinal products on human reproduction and lactation from data to labeling http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003307.pdf