Publications
Teratology Primer, 3rd Edition
Is There a Safe Dose of a Medication Associated with Birth Defects?
Amy L. Inselman and William Slikker Jr., National Center for Toxicological Research, U.S. Food and Drug Administration, Jefferson, Arkansas
During pregnancy, a woman may be hesitant to take prescription medication for fear of harming her unborn fetus. However, it is not often feasible to simply exclude health-benefiting medications during pregnancy to reduce risk. If left untreated or unmanaged, chronic conditions including asthma, high blood pressure, epilepsy, and diabetes can harm both the mother and fetus. Therefore, the establishment of safe levels of exposure to possible teratogenic medications is an important area of research to inform counseling and establish intervention strategies for women inadvertently exposed during pregnancy, as well as for determining which drugs are appropriate for use in pregnancy.
Figure 1.
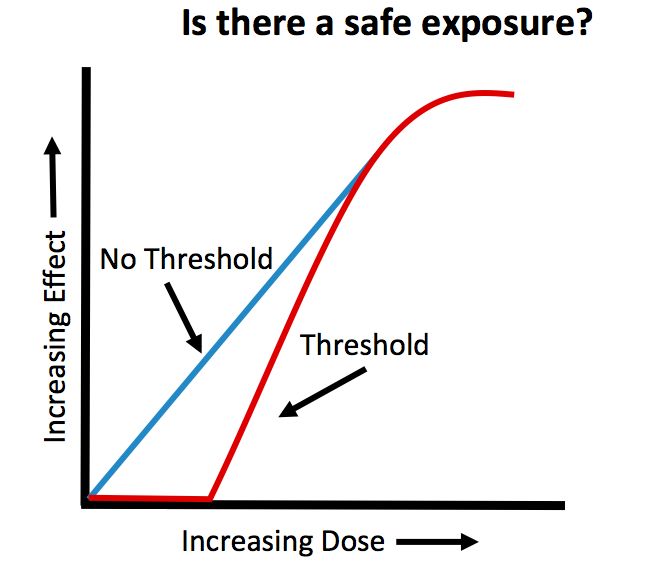
Low doses of compounds may not pose a risk to the embryo or fetus. However, any agent may be harmful if the exposure concentration is high enough. The idea that safe doses exist is based on the concept of the “threshold dose.” The threshold dose is defined as the lowest dose of a drug (or environmental agent) that generates an adverse response (i.e. birth defect). This means for exposures that occur below the threshold level, the risks of adverse effects are equal to those in non-exposed individuals. In other words, there is no risk for irreversible effects or death at doses below the threshold concentration. An example of a compound that demonstrates effects consistent with a threshold is thalidomide. Thalidomide was originally given to treat multiple ailments including morning sickness. This now recognized human teratogen produces severe limb defects when administered at a 50 mg dose early during pregnancy, but a much lower dose of 0.5 mg given at the same time of pregnancy might produce no observable teratogenic effects. Unfortunately, the dose-response relationships for developmental toxicity of most agents in humans are not known, as the drugs for a given condition are usually prescribed over a narrow range and the lowest dose of pharmaceutical products is limited by effectiveness. However, several studies on valproic acid, a drug used to prevent seizures and for treatment of bipolar disorder, have shown that doses above 1000 mg per day, which may be within the therapeutic dose range for some women, are associated with an increased risk of birth defects compared to doses less than 1000 mg/day.
The concept of the threshold dose also assumes that embryotoxicity is dependent on multicellular injury that overwhelms the repair mechanisms of the embryo. Since repair mechanisms exist and are functional during embryonic development, it seems reasonable that minor insults from teratogenic exposures below the threshold dose could be overcome. Indeed, apoptosis, or programmed cell death, is a normal and necessary part of development. Excessive cellular injury, however, could instead result in widespread cell death, producing malformations. Maternal hyperthermia, exposure to ionizing radiation, and retinoic acid have all been shown to induce structural malformations through abnormal apoptotic processes at sufficiently high exposure levels.
While the concept of a threshold dose is widely acknowledged, some scientists disagree and favor a continuum model of probability for the risk of developing malformations. Under a continuum model, there is no safe level of exposure, regardless of how small the exposure may be. Essentially, effects can occur at any dose. Only one third of the malformations that occur in humans have identified genetic or environmental causes. Most birth defects are categorized as spontaneous malformations, meaning their cause is unknown. Therefore, if internal factors can cause spontaneous malformations and exposure to a drug (or environmental cue) also affects the same internal factor, it is not unreasonable to suggest that even low exposures from the environment (or drugs) could tip the balance, resulting in birth defects in some individuals. As an example, consider a drug that alters vitamin A concentrations within embryos. Both too little and too much vitamin A can lead to birth defects. Thus, a drug that either increases or decreases the concentrations of vitamin A within embryos might tip the balance, potentially leading to a malformation.
If the relationship between the dose of a compound and its response is continuous, can there ever be a safe level of exposure? While there is never 100% certainty, using the relationship between dose and outcomes from animal studies or from human cases of exposure, the uncertainty surrounding the risk of fetal malformation can be reduced by administering lower doses of the drug, assuming those doses are effective. Safe doses can be estimated by using safety factors, reducing problematic doses by multiples, usually of 10.
The route and timing of exposure are also critical factors in determining the teratogenicity of a drug. The timing of exposure influences what structures or organ systems are most likely to be affected during development, along with the severity of the malformations. Reducing or eliminating exposures during the most sensitive windows of development is one way risks can be further lowered. In general, the most sensitive period of embryonic development is during organ formation or organogenesis (approximately weeks 3-8 in humans). At this stage of development, the embryo is at the greatest risk for major structural malformations. However, exposures occurring outside of this sensitive window (weeks 9-38) can also produce physiological defects and structural abnormalities. It is important to recognize that the critical period is not the same for all drugs or other agents. Some agents act only during a very small window of development (i.e. organ formation), while others have a much broader range. For example, exposure to thalidomide produces limb malformations when exposure occurs during the first trimester of pregnancy, while the teratogenic effects of the angiotensin-converting enzyme inhibitors used to manage blood pressure occur during the second two-thirds of pregnancy. Exposures to warfarin, ionizing radiation and methyl mercury also are known to induce birth defects outside of first trimester exposure.
Knowledge of drug mechanisms and targets may also be used to lower risk. Antiepileptic drugs (e.g., valproic acid, carbamazepine, phenytoin) are prescribed to women to control seizures. However, this class of drugs is known to interfere with absorption of folic acid, a B vitamin. Low levels of folic acid early during pregnancy are one risk factor for development of neural tube defects. While all women are advised to consume 0.4 mg of folic acid per day before conception and through the first trimester, women who take antiepileptic medications are recommended to take up to 4 mg a day to lessen the chances of neural tube defects. Thus, understanding the mechanism of action for teratogens allows for intervention strategies that can reduce the potential adverse consequences of exposure.
Quantitative biologically based dose-response models are a newer approach being used to mathematically estimate safer doses. Physiologically based pharmacokinetic (PBPK) models consider the physiological and metabolic changes that occur during pregnancy to help determine not only safe but effective doses. During pregnancy, there are many changes that occur physiologically and metabolically that can affect how a drug is absorbed, distributed, broken down and excreted. These parameters also vary based on the trimester of pregnancy. For example, studies have found that metabolism of drugs including lithium, lamotrigine, indinavir, and carbamazepine are altered during pregnancy. Metabolism may increase for some drugs, causing drug levels to fall below the therapeutic range, putting both the mother and fetus at risk. Conversely, pregnancy can also slow drug metabolism, resulting in higher exposure levels, which also place the fetus at an increased risk for developing birth defects. PBPK models allow for extrapolation across species and consider the routes of administration to further estimate safe exposure levels, reducing risk.
Protecting the embryo or fetus from the effects of toxic exposures requires not only knowledge of the dose and timing of exposure, but also information of the target site and mechanisms by which toxicity occurs. If we know enough about these areas, the safe use of medications during pregnancy is still possible.
Disclaimer: The views presented in this article do not necessarily reflect those of the Food and Drug Administration.
Suggested Reading
Brent RL. Editorial comment: Definition of a teratogen and the relationship of teratogenicity to carcinogenicity. Teratology 1986, 34:359 360.
Brill A, Torchinsky A, Carp H and Toder V. The role of apoptosis in normal and abnormal embryonic development. Journal of Assisted Reproduction and Genetics 1999, 16:512-519.
Gaylor DW, Sheehan DM, Young JF and Mattison DR. Letters: The threshold dose question in teratogenesis. Teratology 1986, 38:389 391.
Ke AB, Rostami-Hodjegan A, Zhao P and Unadkat JD. Pharmacometrics in pregnancy: an unmet need. Annual Review of Pharmacology and Toxicology 2014, 54:53-69.
Ornoy A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reproductive Toxicology 2009, 28:1-10.
Slikker W, Jr. and Gaylor DW. Quantitative models of risk assessment for developmental neurotoxicants. In Handbook of Developmental Neurotoxicology, Slikker W, Jr. and Chang LW, (eds.) San Diego; Academic Press, 1998 pp. 727 732.
Wilson, JG. Current status of teratology. In: Handbook of Teratology, Vol. 1, General Principles and Etiology, Wilson JG and Fraser FC, (eds.) New York; Plenum Press, 1977 pp. 47 74.